Qu’est-ce qu’un bon accompagnement réglementaire pour une start-up ?
Actualites
1. Être bien accompagné, ce n’est pas une option quand on crée un DM
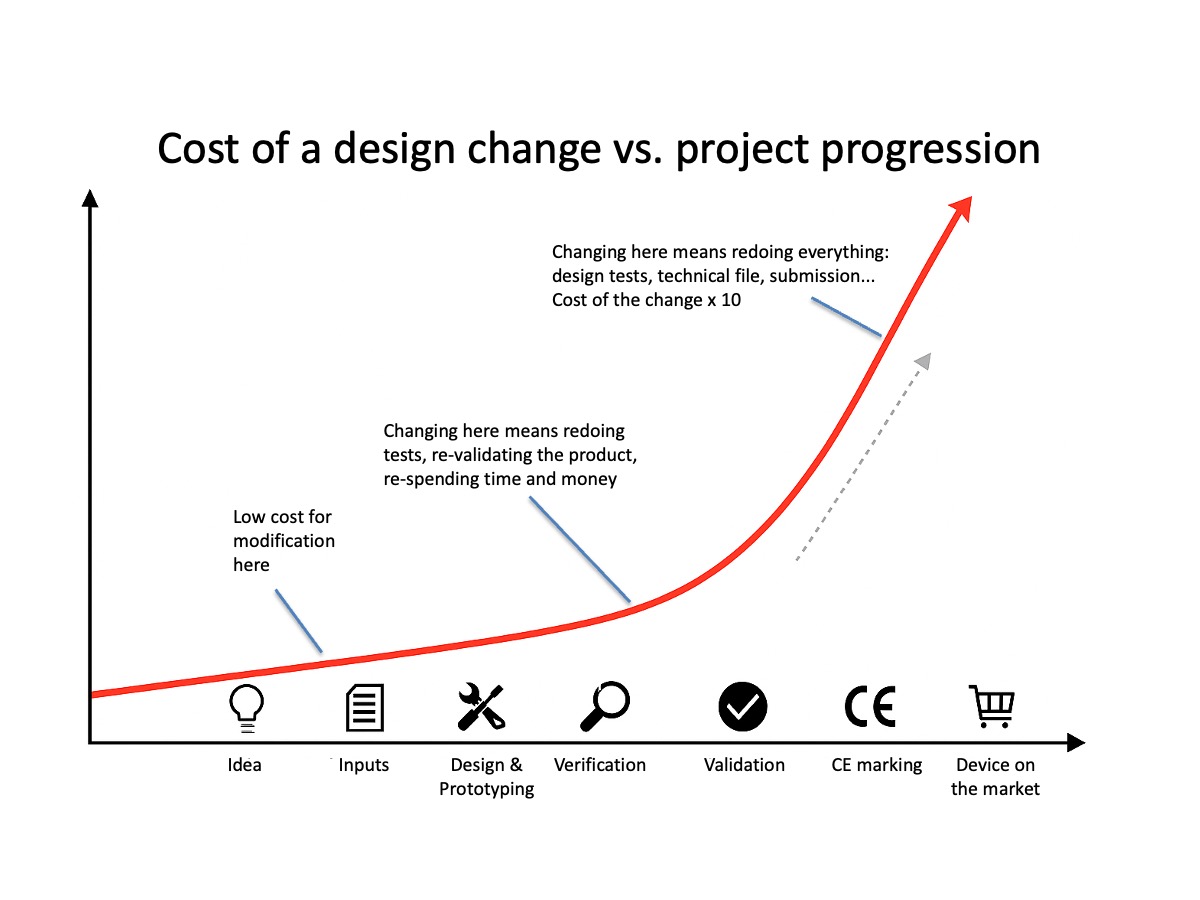
Créer un dispositif médical innovant, ce n’est pas seulement développer un produit qui fonctionne. C’est aussi (et surtout) naviguer dans un environnement réglementaire dense, technique, exigeant.
Le MDR 2017/745 est un texte complexe. Les attentes des organismes notifiés sont élevées. Et les pièges sont nombreux : documentation technique, démonstration clinique, PMS, gestion des risques, classification, UDI, Eudamed… tout s’imbrique. Pour une start-up, la moindre erreur de trajectoire peut coûter des mois – voire la survie du projet.
Dans ce contexte, l’accompagnement réglementaire n’est pas un poste « accessoire ». C’est un investissement stratégique.
2. L’accompagnement réglementaire : un levier stratégique dès la R&D
Une idée peut être brillante… mais si elle est mal positionnée d’un point de vue réglementaire, elle risque de ne jamais voir le marché. Inversement, un accompagnement intelligent permet d’anticiper les bonnes exigences, de faire les bons choix de conception, et de structurer le projet en vue du marquage CE (ou de la FDA).
Un bon conseil réglementaire dès la phase R&D permet de :
- traduire les exigences normatives en critères techniques concrets (design inputs, fonctionnalités, sécurité…),
- anticiper la classe de risque, la règle de classification et les impacts cliniques (investigation ou non ? PMCF ? etc.),
- adapter la stratégie de vérification/validation, en tenant compte du niveau de preuve attendu.
3. Les ingrédients d’un bon accompagnement réglementaire
Un bon consultant réglementaire ne se contente pas de réciter le MDR. Il doit :
- Faire preuve de pédagogie : expliquer les textes sans les caricaturer ni les rendre plus flous qu’ils ne sont.
- Être pragmatique : adapter ses conseils au niveau de maturité de la start-up (tech, clinique, financier).
- Avoir une vision d’ensemble : connecter les points entre R&D, gestion des risques, clinique, qualité, stratégie de dépôt.
- S’impliquer : co-construire la trajectoire, pas juste livrer des documents.
Bref, un bon accompagnement, c’est autant du fond que de la posture. Ce n’est pas “faire à la place de”, c’est “faire grandir” la capacité de l’équipe à comprendre, arbitrer, décider.
4. Ce que CSDmed fait
Chez CSDmed, on n’est pas des chasseurs de livrables.
On accompagne des start-ups qui partent d’un besoin médical, d’une technologie, d’une intuition. Et on les aide à structurer un chemin réglementaire clair, réaliste, cohérent avec leurs moyens et leur ambition.
Ce qui fait la différence :
- Une approche intégrée : réglementaire, qualité, clinique, R&D… tout est connecté.
- Une posture de partenaire : pas d’effet tunnel, on avance ensemble.
- Une adaptation au contexte : accompagnement fractionné si besoin, selon l’avancement ou les financements.
- Une vision internationale : CE, FDA, UKRP, mandataire… on est à l’aise sur plusieurs terrains.
5. Cas pratiques
Cas 1 – Éviter 6 mois de retard grâce à une bonne anticipation
Une start-up en cardiologie prévoyait une étude clinique en France pour valider son DM implantable. Lors de notre revue préliminaire, nous avons identifié un mauvais choix de classe (IIb au lieu de III), basé sur une interprétation incomplète de la règle 8 du MDR.
Conséquence : sans notre intervention, l’étude aurait été lancée sans l’avis d’un organisme notifié, ce qui aurait rendu les données cliniques inutilisables pour le marquage CE.
Nous avons revu la stratégie, contacté l’ON en amont, et permis à la start-up de partir sur des bases solides… et surtout valides.
Cas 2 – Un pivot stratégique pour viser la classe I
Un porteur de projet avait conçu un logiciel de suivi post-opératoire des plaies. Son intuition initiale : “c’est forcément classe IIa, il y a de l’IA et du traitement d’image”.
Après une étude réglementaire sérieuse, nous avons démontré que le DM pouvait, en l’état, être qualifié en classe I, sous certaines conditions. Ce pivot réglementaire a permis d’accélérer la mise sur le marché de 12 à 15 mois, en évitant la soumission à un ON dans un premier temps. Le produit est désormais commercialisé, avec un plan de montée en gamme progressive.
6. Mini FAQ
Est-ce qu’une start-up a vraiment besoin d’un consultant réglementaire dès le début ?
Oui, surtout si elle développe un produit innovant ou mal cadré dans les règles classiques. Un bon conseil en amont évite les erreurs coûteuses ensuite.
Quels sont les risques d’un mauvais accompagnement ?
Sous-classification, stratégie clinique inadaptée, non-conformité du design, mauvaise documentation technique… Les dégâts peuvent être lourds et longs à corriger.
À quel moment faut-il envisager le marquage CE ?
Dès la phase de conception. Le CE n’est pas une étape finale, c’est une trajectoire qui commence dès les premiers choix de design.
Un accompagnement peut-il aussi couvrir la FDA ?
Oui, chez CSDmed, on est à l’aise sur les deux continents. CE et FDA ne s’opposent pas : ils se coordonnent.
Peut-on travailler en plusieurs étapes selon les financements ?
Oui. On sait adapter le rythme et les jalons à la maturité du projet, sans sacrifier la rigueur.
7. Un bon accompagnement, c’est un levier d’agilité, pas un luxe
La réglementation, bien comprise et bien anticipée, peut devenir un accélérateur pour votre projet. Elle vous force à clarifier, à structurer, à prioriser.
Chez CSDmed, on accompagne les start-ups avec sérieux, souplesse et engagement. On connaît la solitude des débuts, les doutes, les arbitrages financiers… et on sait transformer ces contraintes en trajectoires solides.
Vous développez un dispositif médical et vous avez besoin d’un partenaire pour structurer votre démarche réglementaire ?
Écrivez-nous. On verra ensemble comment transformer vos ambitions en réalité.
Ressources associées
- CECP : à quoi doivent être vigilants les fabricants de dispositifs médicaux ?
- Soumission du dossier technique IVDR : ce que les fabricants doivent vraiment soigner (Team-NB, sept. 2025)
- ISO 14971, ISO/TR 24971, IEC 60812 : comment choisir les bonnes méthodes d’analyse des risques ?
- FDA vs MDR : 5 différences qui comptent pour un fabricant de DM