
Post-market surveillance (PMS) sous MDR et IVDR : ce que le guide MDCG 2025-10 change vraiment pour les fabricants
Reglementation des dispositifs medicaux
La post-market surveillance fait partie de ces exigences réglementaires que tous les fabricants savent obligatoires, mais que peu exploitent réellement comme un outil de pilotage. Sous MDR et IVDR, la PMS est pourtant au cœur de la démonstration de conformité sur la durée de vie du dispositif.
Avec le guide MDCG 2025-10, publié le 19 décembre 2025, les autorités européennes apportent enfin une lecture cohérente, structurée et opérationnelle de ce qu’elles attendent concrètement d’un système PMS.
Ce document ne crée pas de nouvelles obligations. Il clarifie, précise et surtout relie entre eux des concepts souvent traités de manière fragmentée par les fabricants.
Que couvre réellement le guide MDCG 2025-10 sur la PMS ?
Le guide MDCG 2025-10 s’applique à l’ensemble des dispositifs médicaux et dispositifs médicaux de diagnostic in vitro, toutes classes confondues. Son objectif est triple :
décrire ce qu’est un système de PMS conforme au MDR et à l’IVDR,
expliciter le contenu attendu d’un plan de PMS,
montrer comment la PMS interagit avec les autres processus du système de management de la qualité.
Le guide ne remplace pas les textes réglementaires et ne fournit pas de modèle clé en main. Il n’entre pas dans le détail de la rédaction des PSUR ou des PMS reports, déjà couverts par le guide MDCG 2022-21. En revanche, il explique comment l’ensemble s’articule et comment les données post-market doivent être utilisées.
C’est précisément ce qui manquait à beaucoup de fabricants.
PMS sous MDR et IVDR : rappel des obligations essentielles
Sous MDR article 83 et IVDR article 78, la PMS est définie comme un système, et non comme un simple livrable documentaire.
Il est essentiel de distinguer clairement :
le système de PMS, intégré au SMQ,
le plan de PMS, qui décrit comment ce système est mis en œuvre pour un dispositif ou une famille de dispositifs,
les rapports issus de la PMS, PMS report ou PSUR selon la classe du dispositif.
La PMS doit être proportionnée à la classe de risque, à la destination du dispositif, à son niveau d’innovation et à son historique. Elle s’applique sur toute la durée de vie du dispositif, depuis la mise sur le marché jusqu’à la fin de vie du dernier exemplaire.
Le guide insiste sur un point souvent mal compris : la PMS n’est pas une activité postérieure au marquage CE, elle est pensée dès la phase de développement.
PMS proactive : ce que les autorités attendent réellement
L’un des apports majeurs du guide MDCG 2025-10 est la clarification de la notion de PMS proactive.
Se limiter à :
enregistrer des plaintes,
traiter des incidents quand ils surviennent,
n’est pas suffisant.
Les autorités attendent que le fabricant organise volontairement la collecte d’informations pertinentes, issues de sources multiples, notamment :
données de vigilance,
incidents (non) graves et effets indésirables,
retours utilisateurs et distributeurs,
littérature scientifique et bases de données publiques,
informations relatives à des dispositifs similaires présents sur le marché.
Cette dernière notion est particulièrement importante. La PMS doit permettre de positionner le dispositif par rapport à l’état de l’art, et pas uniquement par rapport à son propre historique.
Le PMS Plan : contenu attendu et erreurs fréquentes
Le PMS Plan, tel que décrit à l’annexe III du MDR et de l’IVDR, est souvent traité comme une formalité. Le guide MDCG 2025-10 montre au contraire qu’il s’agit d’un document structurant.
Le plan doit notamment préciser :
les méthodes de collecte des données,
les méthodes d’analyse, qualitatives et quantitatives,
les indicateurs suivis et les seuils déclencheurs,
les modalités de trending,
les interfaces avec la vigilance, les CAPA et la gestion des risques,
la place du PMCF ou du PMPF, ou la justification de leur non-applicabilité.
En audit, les non-conformités PMS sont très souvent liées à :
des plans trop génériques,
des seuils non justifiés,
une absence de lien explicite avec la gestion des risques,
une PMS décrite mais jamais réellement exploitée.
Le tableau récapitulatif du guide constitue, de fait, une checklist implicite pour les organismes notifiés.
Le cycle PMS, de la donnée terrain à la décision
Le guide décrit la PMS comme un cycle continu, structuré autour de quatre étapes :
1.collecte des données pertinentes,
2.analyse et consolidation,
3.conclusions sur la sécurité, la performance et le bénéfice-risque,
4.décisions et actions, puis révision du plan PMS si nécessaire.
Ce point est fondamental. Une PMS conforme ne se limite pas à collecter de l’information. Elle doit démontrer la capacité du fabricant à analyser, décider et agir.
Les conclusions de la PMS doivent être tracées, documentées et justifiées, qu’elles conduisent ou non à des actions correctives.
PMS et système de management de la qualité : des interfaces clés
Le guide MDCG 2025-10 insiste fortement sur les interactions entre la PMS et les autres processus du SMQ.
La PMS alimente directement :
la gestion des risques selon l’ISO 14971,
l’évaluation clinique ou des performances,
le PMCF ou le PMPF,
la mise à jour des IFU, de l’étiquetage et de la documentation technique,
le SS(C)P lorsque applicable,
les CAPA et, le cas échéant, les FSCA.
La figure 1 du guide synthétise clairement cette logique. Elle montre que la PMS n’est pas un processus isolé, mais un nœud central du SMQ, au même titre que la gestion des risques ou l’évaluation clinique.
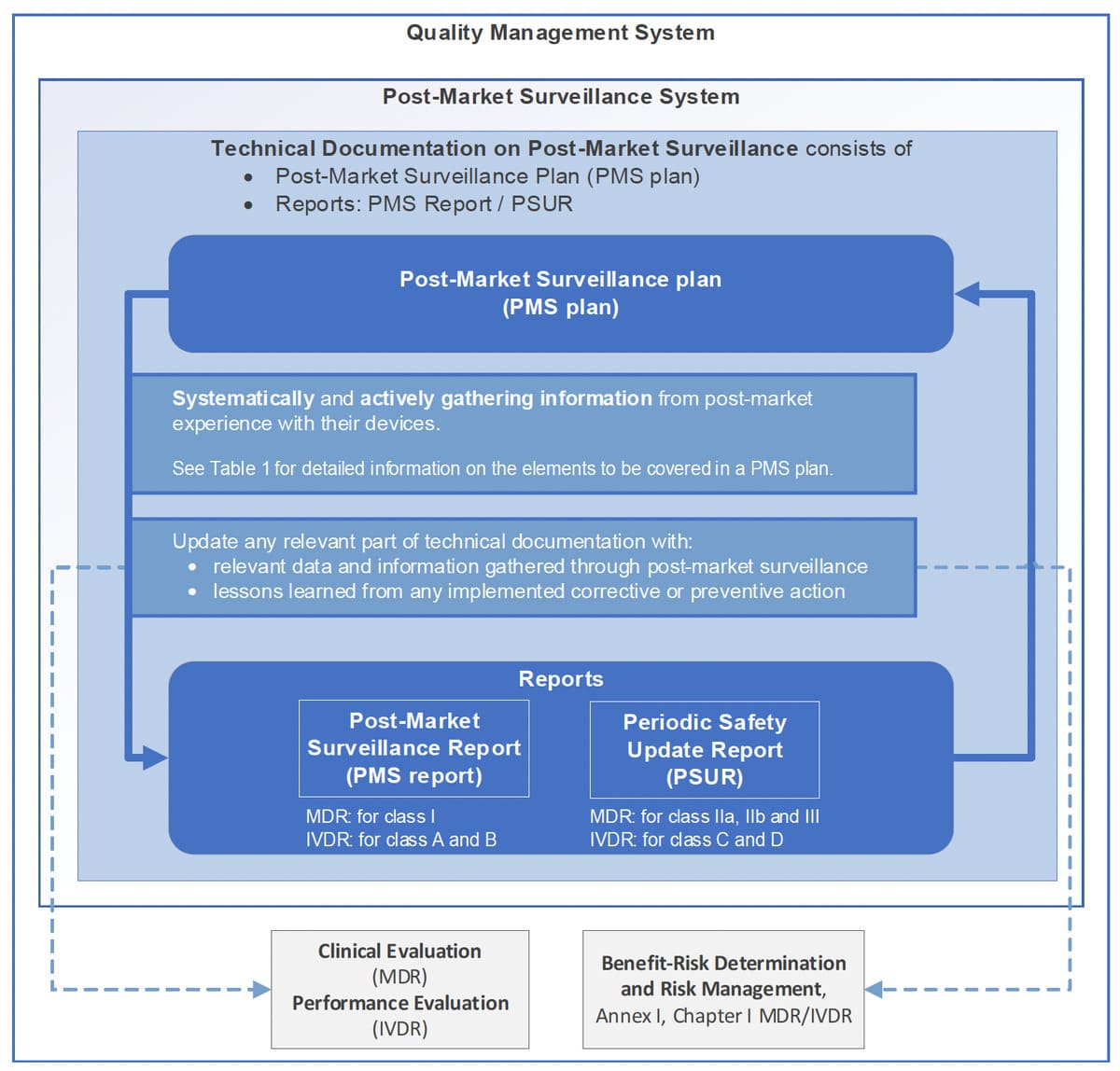
Pour un auditeur, cette cohérence est souvent plus importante que la sophistication des outils utilisés.
Cas pratiques : quand la PMS devient décisionnelle
Le guide illustre son propos par des scénarios concrets.
Dans un cas de dispositif médical implantable, une augmentation de plaintes révèle un risque initialement sous-estimé. La PMS conduit à une mise à jour de la gestion des risques, à des actions correctives, à une FSCA et à une révision de l’évaluation clinique.
Dans un cas de DMDIV, des données post-market mettent en évidence une dérive de performance. La PMS déclenche une CAPA, une communication aux autorités compétentes et une révision du PMPF.
Dans les deux cas, la PMS joue pleinement son rôle : transformer des données terrain en décisions réglementaires structurées.
Ce que ce guide change concrètement pour les fabricants
Pour une start-up, le guide permet de construire une PMS crédible dès le marquage CE, sans surdimensionner inutilement les moyens.
Pour un fabricant déjà certifié, il offre une grille de lecture claire pour sécuriser les audits et corriger les faiblesses récurrentes en PMS.
Pour une organisation multi-dispositifs, il aide à structurer une PMS cohérente, proportionnée et exploitable à l’échelle du portefeuille produit.
Mini FAQ
À partir de quand la PMS doit-elle être active ?
Dès la mise sur le marché du premier dispositif, et de manière continue sur toute sa durée de vie.
Un plan PMS peut-il couvrir plusieurs dispositifs ?
Oui, s’ils partagent une même destination, des caractéristiques de conception et un profil de risque comparable.
Quelle est la frontière entre PMS et PMCF ?
Le PMCF fait partie intégrante de la PMS, lorsqu’il est applicable, et vise à générer des données cliniques spécifiques.
Que regarde un auditeur en priorité ?
La cohérence entre PMS, gestion des risques, évaluation clinique et actions prises.
La PMS peut-elle être externalisée ?
Certaines activités peuvent l’être, mais la responsabilité et l’analyse finale restent celles du fabricant.
Conclusion
Le guide MDCG 2025-10 ne rajoute pas une couche réglementaire supplémentaire. Il explicite ce que les autorités attendent depuis longtemps : une PMS vivante, proportionnée et réellement utilisée pour piloter la sécurité et la performance des dispositifs tout au long de leur cycle de vie.
Dans ce contexte, CSDmed accompagne ses clients, start-ups, fabricants, importateurs et distributeurs de dispositifs médicaux, dans la structuration et la mise en œuvre de systèmes de post-market surveillance conformes au MDR et à l’IVDR. Cette approche s’appuie sur une lecture opérationnelle des exigences réglementaires et sur une intégration cohérente de la PMS avec la R&D, la maîtrise des risques, le système de management de la qualité et la documentation technique.
Contactez-nous pour échanger sur vos enjeux PMS et sécuriser durablement la conformité et la performance de vos dispositifs.
Ressources associées
ISO 14971 and start-ups: how to get started on risk analysis without getting lost
CECP Procedure: What Should Medical Device Manufacturers Watch Out For?