
Post-market surveillance (PMS) under MDR and IVDR: what the MDCG 2025-10 guide really changes for manufacturers
Medical devices regulation
Post-market surveillance (PMS) is one of those regulatory requirements that all manufacturers know is mandatory, but that few actually exploit as a steering tool. Under MDR and IVDR, PMS is at the heart of demonstrating conformity over the lifetime of the device.
With MDCG 2025-10, published on December 19, 2025, the European authorities have finally provided a coherent, structured and operational interpretation of what they expect from a PMS system.
This document does not create new obligations. It clarifies, specifies and, above all, links together concepts that are often dealt with in a fragmented way by manufacturers.
What does MDCG 2025-10 really cover?
MDCG 2025-10 applies to all classes of medical devices and in vitro diagnostic medical devices. Its purpose is threefold:
to describe what a PMS system compliant with MDR and IVDR is,
to explain the expected content of a PMS plan,
to show how PMS interacts with the other processes of the quality management system.
The guide does not replace regulatory texts, nor does it provide a turnkey model. It does not go into detail on the drafting of PSURs or PMS reports, which are already covered by MDCG 2022-21. It does, however, explain how everything fits together and how post-market data should be used.
This is precisely what many manufacturers have been missing.
PMS under MDR and IVDR: reminder of essential obligations
Under MDR article 83 and IVDR article 78, the PMS is defined as a system, and not simply as a documentary deliverable.
It is essential to make a clear distinction between :
the PMS system, integrated into the QMS,
the PMS plan, which describes how this system is implemented for a device or family of devices,
the reports produced by the PMS, PMS report or PSUR, depending on the class of device.
The PMS must be proportionate to the risk class, the purpose of the device, its level of innovation and its history. It applies to the entire life of the device, from the moment it is placed on the market to the end of life of the last unit.
The guide emphasizes a point that is often misunderstood: PMS is not a post-CE-marking activity, but is considered right from the development phase.
Proactive PMS: what the authorities really expect
One of the major contributions of the MDCG 2025-10 guide is the clarification of the notion of proactive PMS.
To be limited to :
recording complaints,
dealing with incidents when they occur,
is not enough.
The authorities expect the manufacturer to voluntarily organize the collection of relevant information from multiple sources, including :
vigilance data,
(non) serious incidents and adverse reactions,
user and distributor feedback,
scientific literature and public databases,
information on similar devices on the market.
This last notion is particularly important. The PMS must make it possible to position the device in relation to the state of the art, and not only in relation to its own history.
The PMS Plan: expected content and common errors
The PMS Plan, as described in Appendix III of the MDR and IVDR, is often treated as a formality. On the contrary, the MDCG 2025-10 guide shows that it is a structuring document.
In particular, the plan must specify :
data collection methods,
qualitative and quantitative analysis methods,
indicators monitored and trigger thresholds,
trending procedures,
interfaces with vigilance, CAPA and risk management,
the place of PMCF or PMPF, or the justification for their non-applicability.
In audits, PMS non-conformities are very often linked to :
overly generic plans
unjustified thresholds,
no explicit link with risk management,
pMS described but never actually used.
The guide's summary table is, in fact, an implicit checklist for notified bodies.
The PMS cycle, from field data to decision
The guide describes PMS as a continuous cycle, structured around four stages:
1.collection of relevant data,
2.analysis and consolidation,
3.conclusions on safety, performance and benefit-risk,
4.decisions and actions, then revision of the PMS plan if necessary.
This point is fundamental. A compliant PMS does more than simply collect information. It must demonstrate the manufacturer's ability to analyze, decide and act.
PMS conclusions must be traced, documented and justified, whether or not they lead to corrective action.
PMS and quality management system: key interfaces
Guide MDCG 2025-10 strongly emphasizes the interactions between the PMS and other QMS processes.
The PMS feeds directly into :
iSO 14971 risk management,
clinical or performance evaluation,
pMCF or PMPF,
iFU, labeling and technical documentation updates,
sS(C)P when applicable,
cAPAs and, where applicable, FSCAs.
Figure 1 of the guide clearly summarizes this logic. It shows that the PMS is not an isolated process, but a central node of the QMS, in the same way as risk management or clinical evaluation.
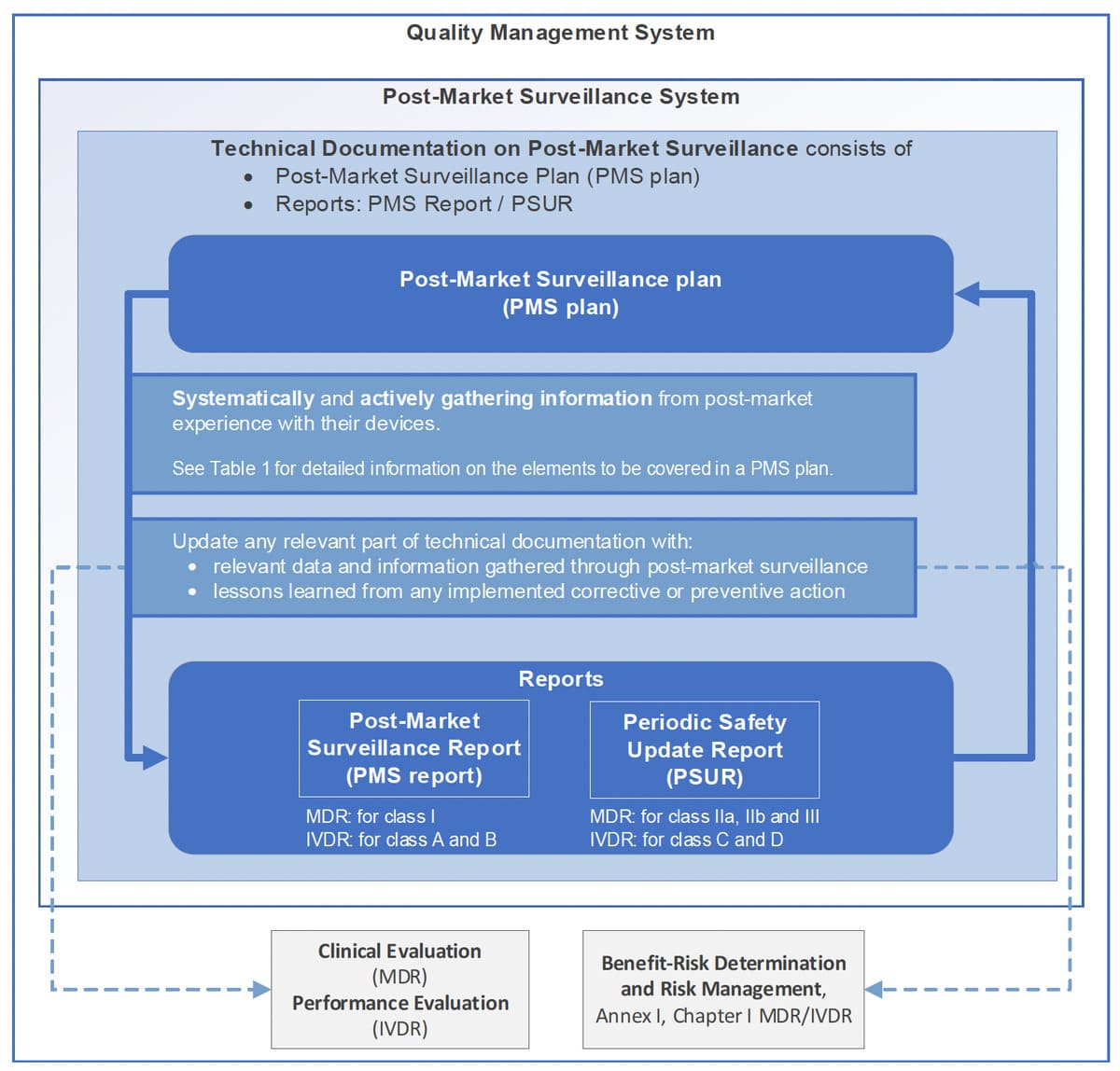
For an auditor, this coherence is often more important than the sophistication of the tools used.
Case studies: when PMS becomes a decision-making tool
The guide illustrates its point with concrete scenarios.
In the case of an implantable medical device, an increase in complaints reveals an initially underestimated risk. The PMS leads to an update of risk management, corrective actions, an FSCA and a review of the clinical evaluation.
In the case of a DMDIV, post-market data highlight a performance drift. The PMS triggers a CAPA, a communication to the competent authorities and a review of the PMPF.
In both cases, the PMS plays its role to the full: transforming field data into structured regulatory decisions.
What this guide means in concrete terms for manufacturers
For start-ups, the guide enables them to build a credible PMS right from the CE marking stage, without unnecessarily overstretching their resources.
For an already-certified manufacturer, it provides a clear framework for securing audits and correcting recurring PMS weaknesses.
For a multi-device organization, it helps to structure a PMS that is coherent, proportionate and usable across the entire product portfolio.
Mini FAQ
When should PMS be active?
As soon as the first device is placed on the market, and continuously throughout its life cycle.
Can a PMS plan cover several devices?
Yes, if they share the same purpose, design characteristics and risk profile.
What is the boundary between PMS and PMCF?
The PMCF is an integral part of the PMS, when applicable, and aims to generate specific clinical data.
What does an auditor look for first?
Consistency between PMS, risk management, clinical assessment and actions taken.
Can PMS be outsourced?
Certain activities can be outsourced, but responsibility and final analysis remain with the manufacturer.
Conclusion
MDCG 2025-10 does not add another layer of regulation. It spells out what the authorities have long been waiting for: a living, proportionate PMS that is actually used to steer the safety and performance of devices throughout their lifecycle.
In this context, CSDmed supports its customers - start-ups, manufacturers, importers and distributors of medical devices - in structuring and implementing post-market surveillance systems that comply with the MDR and IVDR. This approach is based on an operational reading of regulatory requirements, and on the coherent integration of PMS with R&D, risk management, the quality management system and technical documentation.
Contact us to discuss your PMS challenges and secure the long-term compliance and performance of your devices.
Related resources
ISO 14971 and start-ups: how to get started on risk analysis without getting lost
CECP Procedure: What Should Medical Device Manufacturers Watch Out For?