
Nouvelle version: MDCG 2021-6 Rev. 1 - Questions et réponses concernant l'investigation clinique
Reglementation des dispositifs medicaux
🫵🏻 Nouvelle version aujourd'hui : MDCG 2021-6 Rev. 1
Règlement (UE) 2017/745 Questions et réponses concernant l'investigation clinique
- L'annexe III a été ajoutée pour les produits combinés.
- Le parcours réglementaire de l’annexe I a été légèrement mis à jour.
- Et de nombreuses questions ont été ajoutées ou mises à jour.
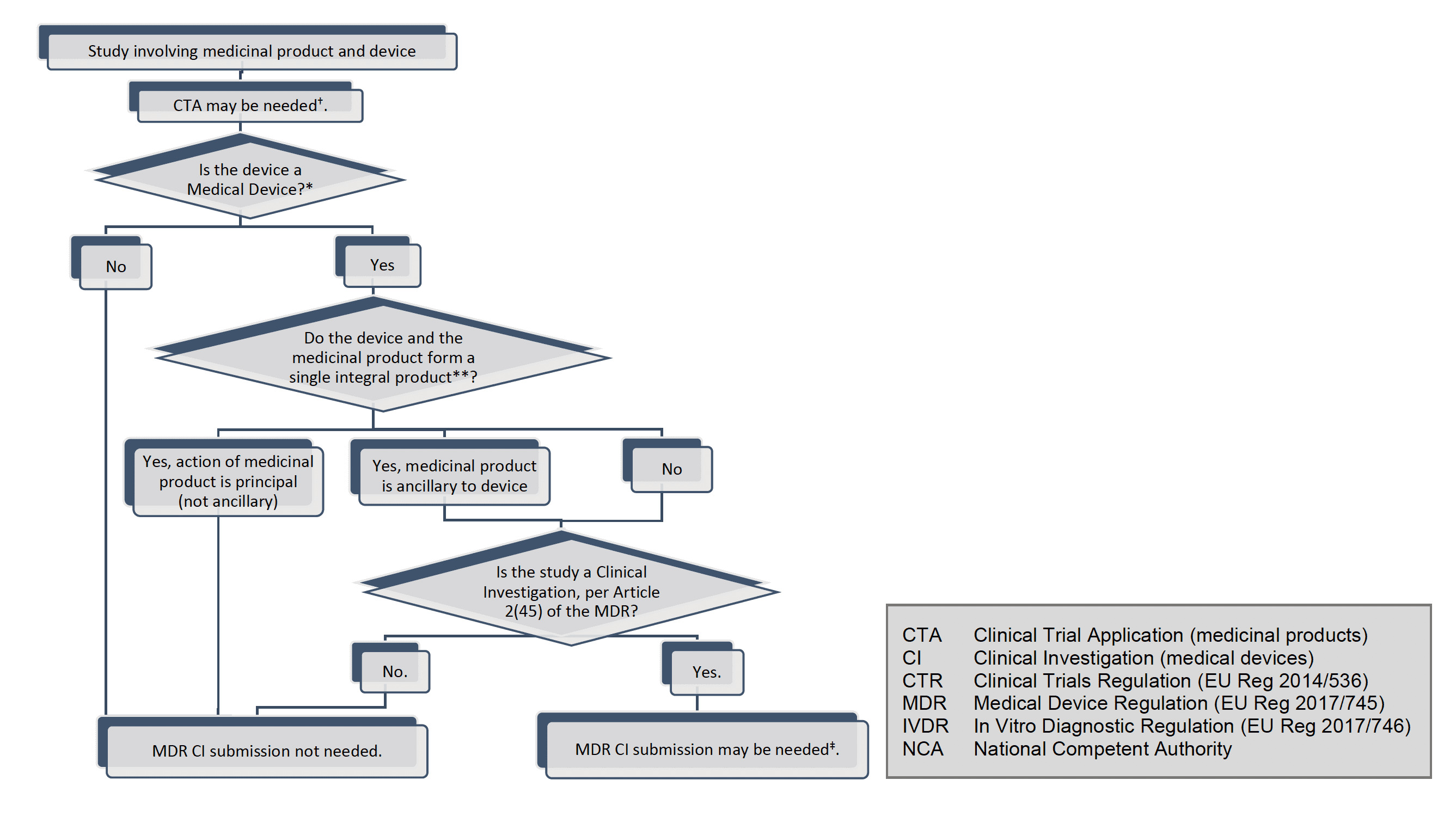
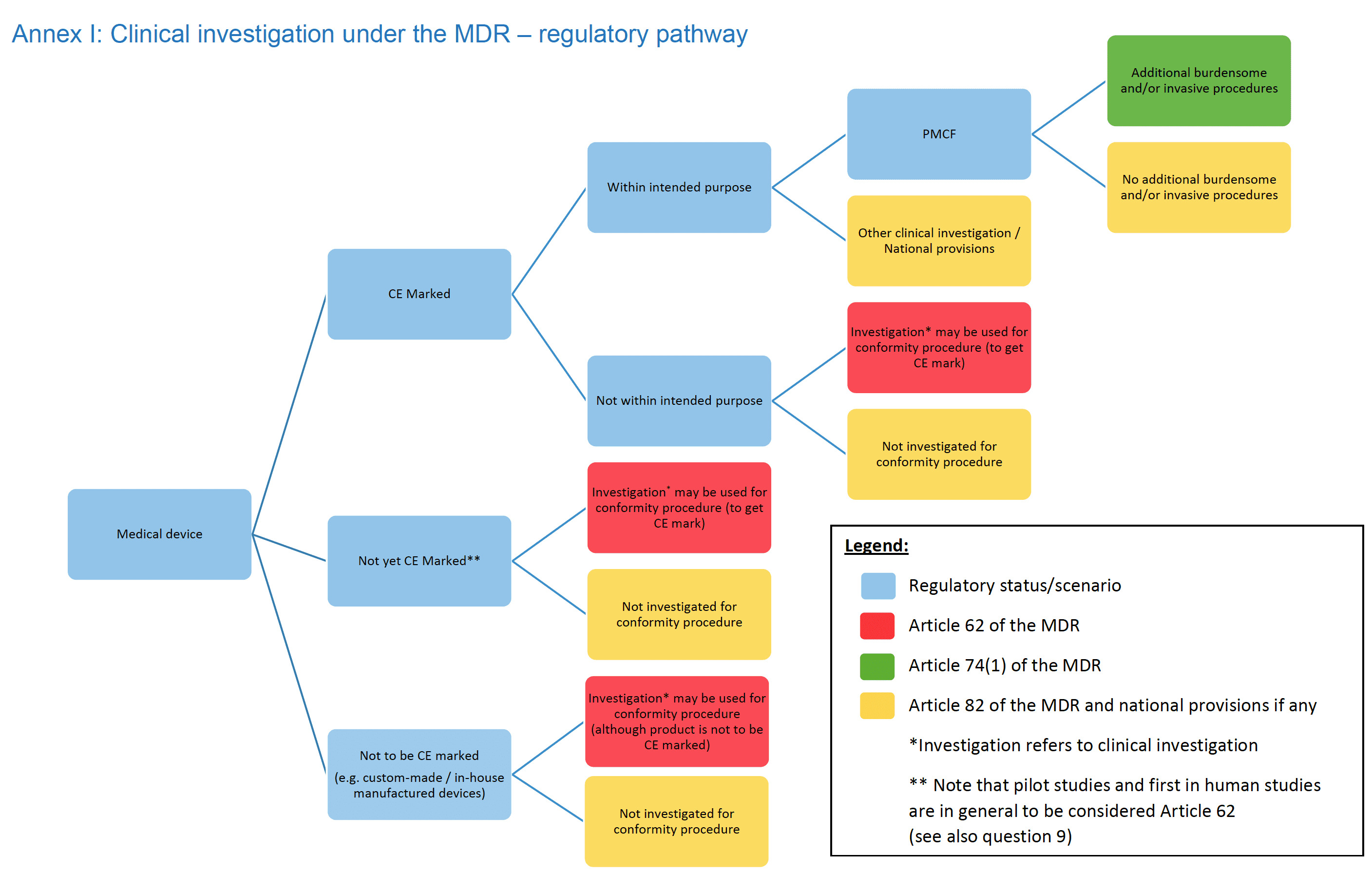
Introduction
Ce document est destiné aux promoteurs d'investigations cliniques sur des dispositifs menées dans le cadre du règlement (UE) 2017/745 (MDR). Ce document pourra être complété en temps utile par d’autres questions et réponses. Dans tout le présent document, le terme « appareil » est utilisé avec la même signification que dans le MDR, c'est-à-dire qu'aux fins du MDR, les dispositifs médicaux, les accessoires pour dispositifs médicaux et les produits répertoriés à l'annexe XVI du MDR et auxquels le MDR s'applique, doivent ci-après dénommés ꞌdispositifsꞌ.
En outre, le terme « investigation clinique » est utilisé partout dans le même sens que dans l'article 2(45) du MDR, c'est-à-dire « toute investigation systématique impliquant un ou plusieurs sujets humains, entreprise pour évaluer la sécurité ou les performances d'un dispositif ».
Si l'investigation clinique est clairement définie dans le MDR, il est parfois également nécessaire de mentionner qu'il existe des études qui ne répondent pas à cette définition, pour tracer la frontière entre le moment où les dispositions du chapitre VI et de l'annexe XV du MDR s'appliquent, et celles où elles ne le sont pas. À cette fin, le terme plus large « étude clinique » est utilisé et couvre, aux fins de ces lignes directrices, les études réalisées dans le cadre de la recherche médicale impliquant des humains et comprend les essais cliniques de médicaments, les investigations cliniques de dispositifs et les études de performances cliniques de tests in vitro. dispositifs de diagnostic, ainsi que d'autres études dans un cadre clinique où des produits tels que des médicaments ou des dispositifs ne sont pas nécessairement impliqués du tout.
De plus, le promoteur doit être conscient que le MDR ne précise pas de détails sur l'examen éthique des investigations cliniques. Il est donc nécessaire de vérifier les exigences nationales en matière de soumission au comité d'éthique et, le cas échéant, de s'assurer que les comités d'éthique et les autorités compétentes ont accès aux mêmes versions des documents mis à jour.
Nous sommes à votre service
Depuis l’arrivée du Règlement 2017/745, de nouvelles exigences sont demandées et font partie de la documentation à présenter à l’organisme notifié (par exemple revue de l’évaluation clinique poussée, procédure de PMS, résultats de CQ issus de la validation des produits, preuves de compétences du personnel, etc.).
CSDmed apporte son expertise et une approche méthodique à ses clients, start-ups, fabricants, importateurs et distributeurs de dispositifs médicaux, grâce à une équipe d’experts et de consultants spécialisés, qui pourront traiter la transition MDR dans son entièreté.
🔗 Contactez-nous et découvrez comment nous pouvons vous aider.