
MDCG 2023-7 Guidance on exemptions from the requirement to perform clinical investigations pursuant to Article 61(4)-(6) MDR and on sufficient levels of access to data needed to justify claims of equivalence
Medical devices regulation
MDCG 2023-7 Guidance on exemptions from the requirement to perform clinical investigations pursuant to Article 61(4)-(6) MDR and on ‘sufficient levels of access’ to data needed to justify claims of equivalence
This guidance is intended to clarify the exemptions from the requirement to perform clinical investigations, and associated conditions related to the demonstration of equivalence, for implantable and class III medical devices to be placed on the European market. It also provides examples and considerations relevant to the demonstration of “sufficient levels of access to the data” per Annex XIV Section 3.
What can we find in this Guidance ?
The document elaborates on specific conditions under which clinical investigations for medical devices might be exempted. For example, if a new medical device is similar to an existing, approved device, the manufacturer can claim equivalence and potentially bypass additional clinical trials. This equivalence must be justified with data demonstrating similarity in technical, biological, and clinical aspects. It includes scenarios like a manufacturer comparing their new device to an existing one they produce or to a competitor's device, provided they have adequate access to the necessary data. The document underscores the importance of thoroughly justifying any claim of equivalence, ensuring that patient safety and device effectiveness are not compromised by the exemption from clinical trials.
4 scenarios
The MDCG 2023-7 guideline describes in Table 1 four scenarios in which exemptions from clinical investigations are possible:
Case 1: Indents 1-3 of Article 61(4):
- The device under evaluation (DUE) has been designed by modifying a device already marketed by the same manufacturer.
- Equivalence is demonstrated between the DUE and the manufacturer’s ED (Equivalent Device) in accordance with Section 3 of Annex XIV; demonstration of equivalence has been endorsed by the notified body. For further guidance on the demonstration of equivalence, please refer to MDCG 2020-5.
- The clinical evaluation of the marketed device is sufficient to demonstrate conformity of the modified device with the relevant safety and performance requirements.
- PMCF plan is appropriate and includes post market studies to demonstrate the safety and performance of the DUE.
Case 2: Article 61(6)(a):
- DUE has been lawfully placed on the market or put into service in accordance with Directive 90/385/EEC or Directive 93/42/EEC.
- The clinical evaluation is based on sufficient clinical data.
- The clinical evaluation is in compliance with the relevant product-specific CS for the clinical evaluation of that kind of device, where such a CS is available.
Case 3: Article 61(6)(b):
- DUE is one of the listed types of devices: “sutures, staples, dental fillings, dental braces, tooth crowns, screws, wedges, plates, wires, pins, clips or connectors”.
- The clinical evaluation is based on sufficient clinical data.
- The clinical evaluation is in compliance with the relevant product-specific CS for the clinical evaluation of that kind of device, where such a CS is available.
Case 4: Article 61(5):
- Equivalence is demonstrated between the DUE and the other manufacturer’s ED in accordance with Section 3 of Annex XIV.
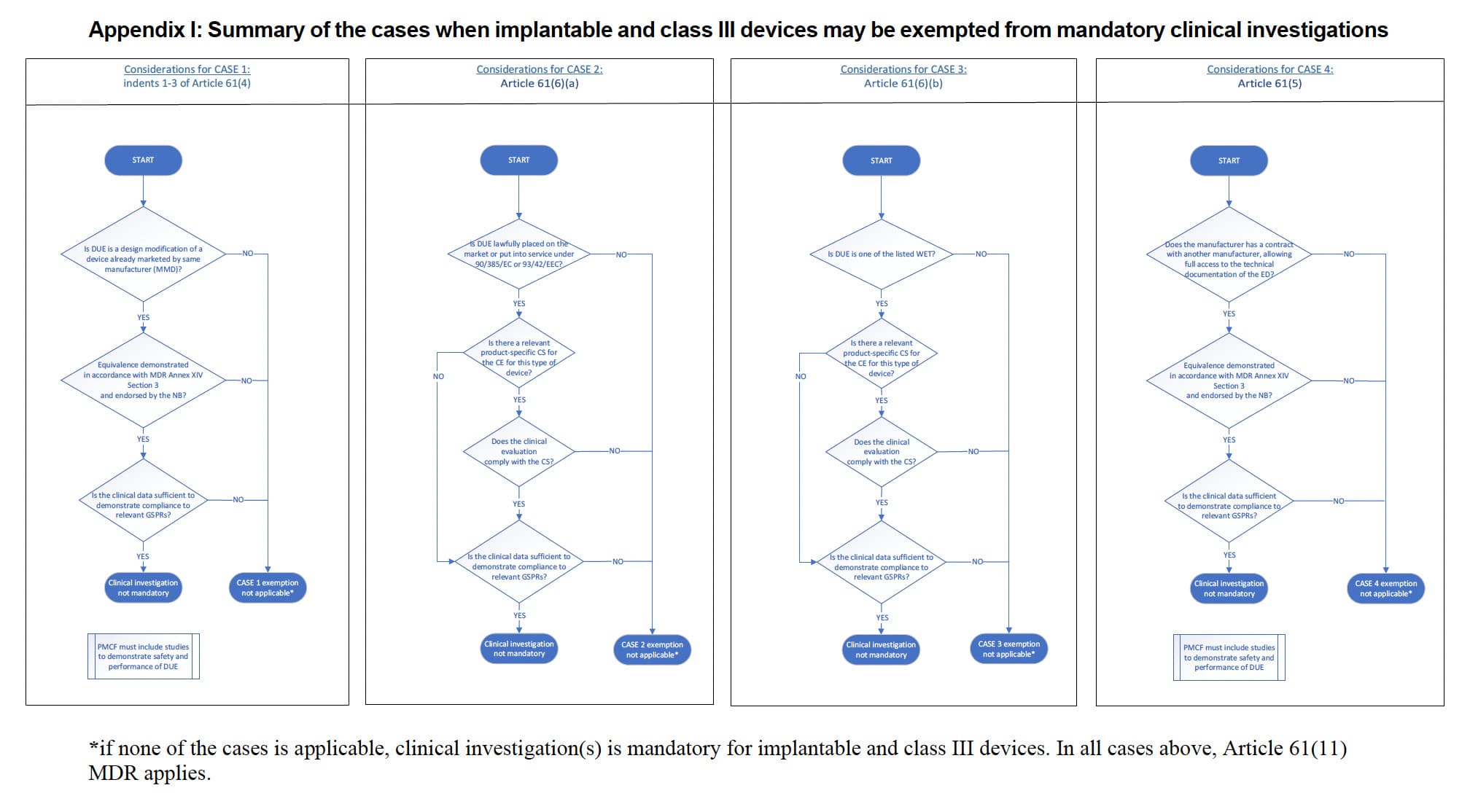
The document includes two annexes:
1️⃣ Appendix I summarizes cases where implantable and class III devices may be exempted from mandatory clinical investigations. It details specific scenarios under which these exemptions apply, helping manufacturers determine if their device qualifies for exemption.
2️⃣ Appendix II presents a hierarchy of levels of access to data regarding clinical, technical, and biological characteristics for demonstrating equivalence. This appendix provides examples and suggests a hierarchy related to the level of access to data, indicating potential limitations and how they might be addressed.
These annexes are crucial for understanding the conditions under which clinical investigations for certain medical devices can be exempted and how to justify claims of equivalence based on the level of data access.
Link to the guide below. Enjoy 💕
We are at your service
Since the arrival of Regulation 2017/745, new requirements are requested and form part of the documentation to be presented to the notified body (e.g. review of the in-depth clinical evaluation, PMS procedure, QC results from validation products, proof of staff skills, etc.).
CSDmed brings its expertise and a methodical approach to its clients, start-ups, manufacturers, importers and distributors of medical devices, thanks to a team of specialized experts and consultants, who will be able to address the MDR transition in its entirety.
🔗 Contact us and find out how we can help you.