Implementing Regulation (EU) 2022/2346 and (EU) 2023/1194 of the arrangements in Annex XVI of MDR 2017/745 - risk management and transitional provisions
Medical devices regulation
Certain devices without an intended medical purpose are nevertheless subject to regulation MDR 2017/745 relating to medical devices. They are considered to have risks comparable to medical devices and are listed in Annex XVI of the MDR.
Implementing Regulation (EU) 2022/2346 amended by Implementing Regulation (EU) 2023/1194 defines common specifications for these non-medical products. The transitional arrangements and risk management requirements are described there.
Reminder of the expectations of MDR 2017/745 regarding Annex XVI products
For devices listed in Annex XVI, the general safety requirements established in points 1 and 8 of Annex I, Chapter I, are interpreted as meaning that the device, used under normal conditions and in accordance with its intended purpose, presents no risk or a risk which is not greater than the maximum acceptable risk linked to the use of the product, so as to guarantee a high level of protection of the safety and health of people.
General requirements
The general requirements in terms of Risk Management and user information are described in Annex I of the Regulation (EU) 2023/1194
⚠️ Risk management
The risk management process is recalled (and it remains very close to the principles of ISO 14971):
- risk management (RM) planning;
- References and description of the devices
- List of risk management activities to be carried out
- Specification of the phases of the device life cycle covered by the RM
- Roles and responsibilities
- Risk acceptability criteria*
- Criteria for collecting information in production and post-production
- hazard identification and risk analysis;
- Under normal conditions of use and in the event of reasonably foreseeable misuse.
- Estimation of the severity and probability of occurrence of damage
- Risk Assessment;
- Determine whether risks are acceptable
- risk management and assessment of residual risks;
- List of risk control measures and evaluation of their effectiveness
- List of residual risks after risk reduction
- Risk management in the following order of preference:
- a) the integration of security into the design;
- b) the integration of safety into manufacturing;
- c) protective measures in the device itself or within the manufacturing process;
- d) safety information and, where applicable, user training.
- review of risk management;
- Check that risk management procedures have been respected
- The overall residual risk is acceptable, and the risks eliminated or reduced as much as possible.
- production and post-production activities.
- Information collection and review process
* Concerning the acceptability criteria, here is what the implementing regulations specify:
Manufacturers take into account that all risks, including risks associated with surgical intervention, must be eliminated or reduced as much as possible. If the undesirable side effects are of a transient nature and do not require medical or surgical intervention to prevent a life-threatening illness, permanent impairment of an anatomical function or permanent alteration of an anatomical structure, the residual risks can be considered acceptable. If one or more of the conditions set out in this section are not met, the manufacturer provides justification to demonstrate the acceptability of the risks.
📄 Safety information
- As the destination is non-medical, the information must not contain any claim of clinical benefit
- Label
- Bears the words “non-medical destination:” followed by a description of this non-medical destination.
If possible, information relating to user categories;
the expected performance of the device;
the risks arising from the use of the device.
- Instructions for use
- Includes information relating to user categories;
Description of the expected performance of the device (what non-medical effect can be expected from the use of the device);
Description of the residual risks of the device, including associated control measures, presented in a clear and easily understandable manner, so that the consumer can make an informed decision whether to be treated with this device or to have it implanted or use it in any other way;
The expected lifespan or expected resorption period of the device as well as any necessary follow-up;
Reference to any harmonized standard and any common specification that applies to the device.
Specific requirements
The specific requirements for each type of product listed in Annex XVI of MDR 2017/745 are listed in Annexes II to VII of Regulation 2022/2346.
Transitional provisions for non-medical devices
The rules are defined in Article 2 of regulation 2022/2346, and were amended by amending regulation 2023/1194.
In essence, here is what you need to remember:
Terms and conditions
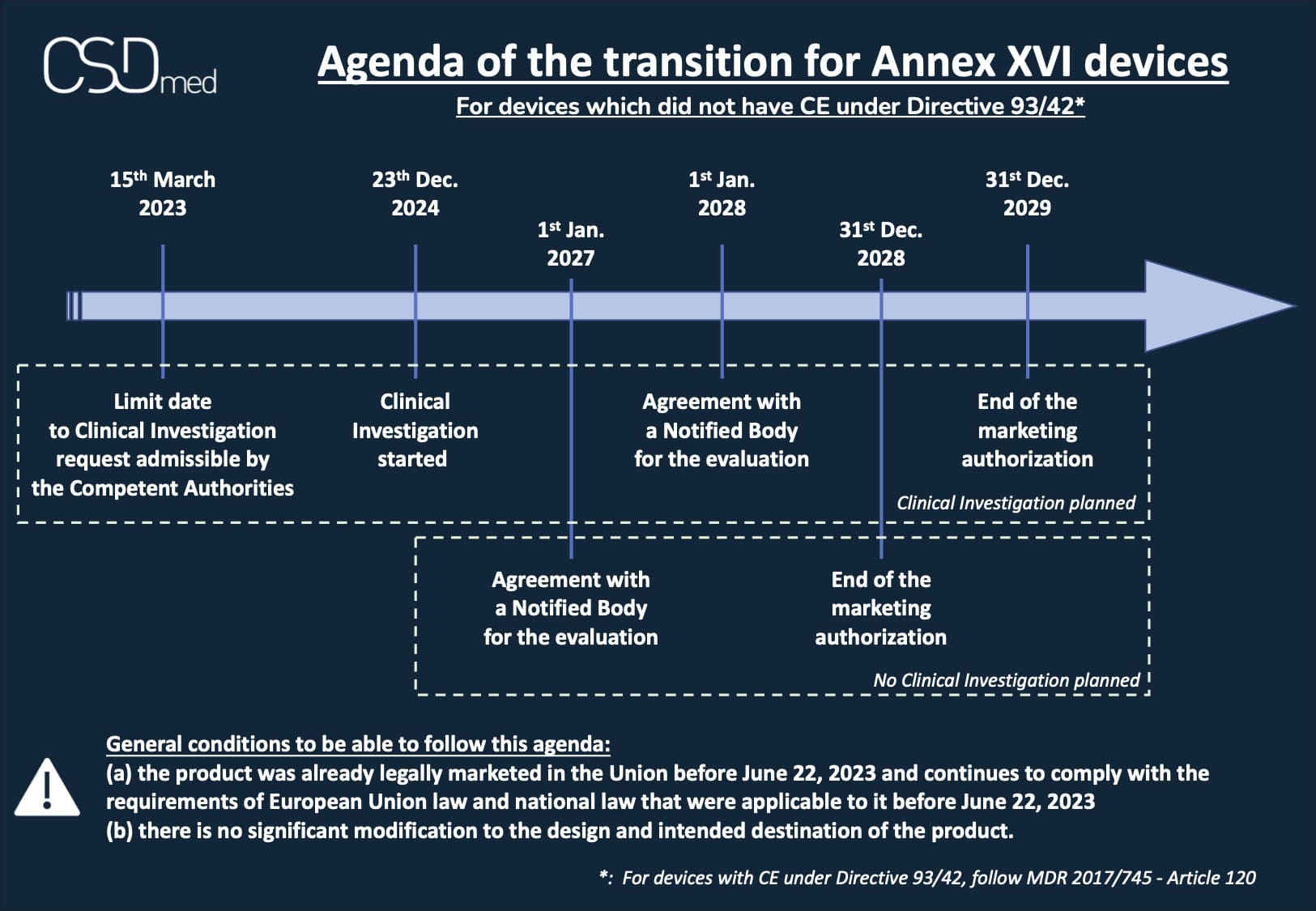
- the product was already legally marketed in the Union before June 22, 2023 and continues to comply with the requirements of European Union law and national law that were applicable to it before June 22, 2023
- there is no significant modification to the design and intended destination of the product.
Devices which did not have CE under Directive 93/42
Timeline if there is a Clinical Investigation (CI) planned (at the latest):
- June 22, 2024: CI request admissible by the Competent Authorities (Article 2 - 1 of EU 2022/2346)
- December 23, 2024: the CI started (Article 2 - 1 of EU 2022/2346)
- January 1, 2028: agreement with a Notified Body (NB) for the evaluation (Article 2 - 1 of EU 2022/2346 amended)
- December 31, 2029: end of the marketing authorization (Article 2 - 1 of EU 2022/2346 amended)
Schedule if there is no CI planned (at the latest):
- January 1, 2027: agreement with a Notified Body (NB) for the evaluation (Article 2 - 2 of EU 2022/2346 amended)
- December 31, 2028: end of the marketing authorization (Article 2 - 2 of EU 2022/2346 amended)
Devices which had a CE under Directive 93/42
Calendar identical to that of MDR Article 120, here is the reminder:
- May 26, 2021: application of the MDR to class I devices or any new devices
- March 20, 2023: agreement with a Notified Body (NB), or Competent Authority, or CE under MDD
- May 26, 2024: request from a Notified Body (ON) and SMQ in agreement with the MDR
- September 26, 2024: agreement with a Notified Body (ON) for the evaluation
- May 26, 2026: end of the marketing authorization for custom implantable class III devices
- December 31, 2027: end of the marketing authorization for class III and IIb implantable devices
- December 31, 2028: end of the marketing authorization for non-implantable class IIb and IIa devices
You can consult our article on The calendar according to amending regulation 2023/607.
We are at your service
Since the arrival of Regulation 2017/745, new requirements are requested and form part of the documentation to be presented to the notified body (e.g. review of the in-depth clinical evaluation, PMS procedure, QC results from validation products, proof of staff skills, etc.).
CSDmed brings its expertise and a methodical approach to its clients, start-ups, manufacturers, importers and distributors of medical devices, thanks to a team of specialized experts and consultants, who will be able to address the MDR transition in its entirety.
🔗 Contact us and find out how we can help you.